An open-source, all-atom engine for drug discovery
OpenDDE turns co-folding into a scalable engine for structure prediction, design, and optimization — modeling proteins, nucleic acids, and small molecules in one all-atom system.
Co-folding, at engine scale
Modern drug discovery rarely asks about a single molecule in isolation. It asks how a candidate binds, how a complex assembles, and how to reshape that interaction toward a therapeutic goal. OpenDDE is built for exactly this: an all-atom biomolecular foundation model that co-folds every component of a system together, rather than predicting structures one chain at a time.
A single input describes the full assembly — protein chains, nucleic acids, ligands, ions, and any explicit covalent bonds — and OpenDDE resolves the joint structure at atomic resolution. The same backbone that predicts structure also drives design and optimization, turning what was a prediction tool into a scalable engine you can iterate against.
Three ideas inside OpenDDE
OpenDDE is not a structure predictor with a design mode bolted on. Three choices — how it reasons, how it is supervised, and how it is framed — let a single model fold, reason about, and generate biomolecular structure.
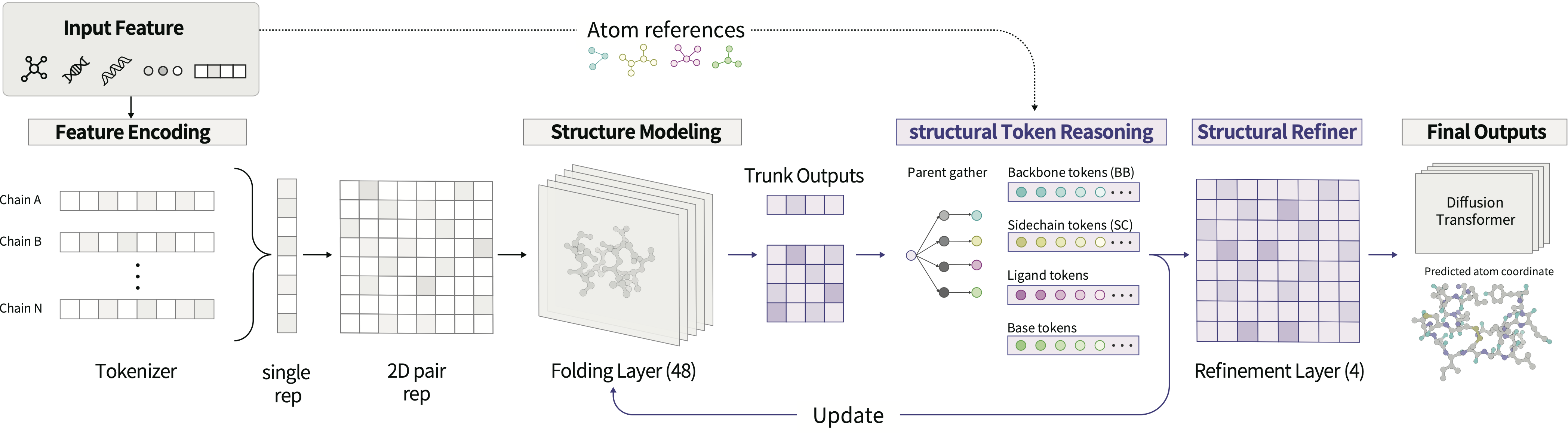
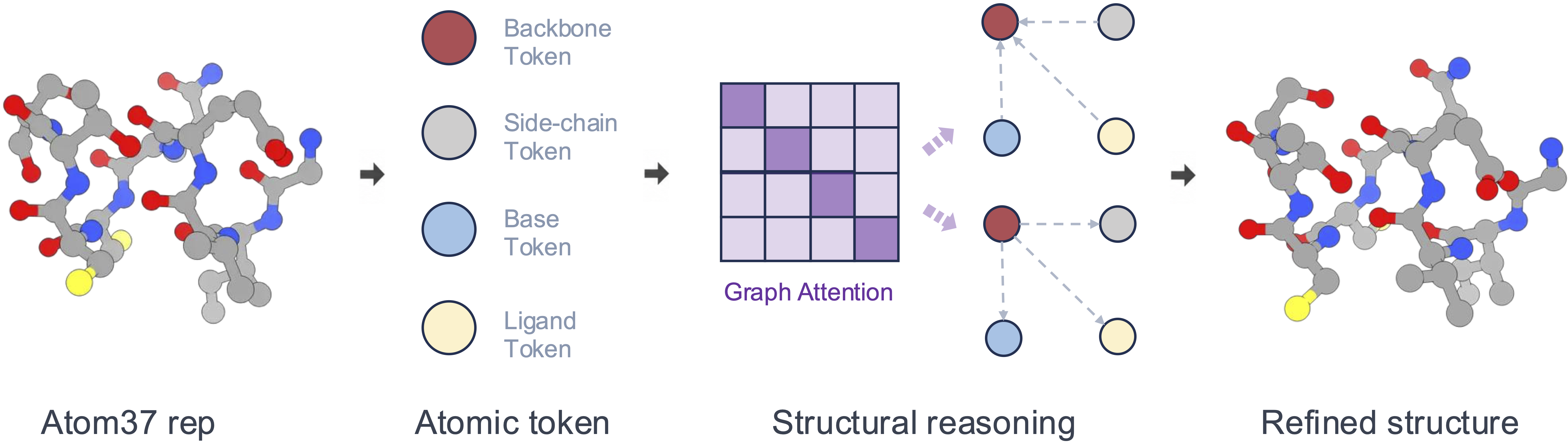
Atomic latent reasoning
OpenDDE reasons from coarse to fine. A Pairformer-style trunk first builds residue-level single and pair representations, then expands every residue token into chemically explicit structural tokens — protein backbone and side chain, nucleic-acid backbone and base, ligand, and single atoms. Before any coordinates are generated, a structural refiner runs several rounds of latent-space reasoning over these tokens, sharpening local geometry, chemical environment, and cross-molecular interfaces up front.
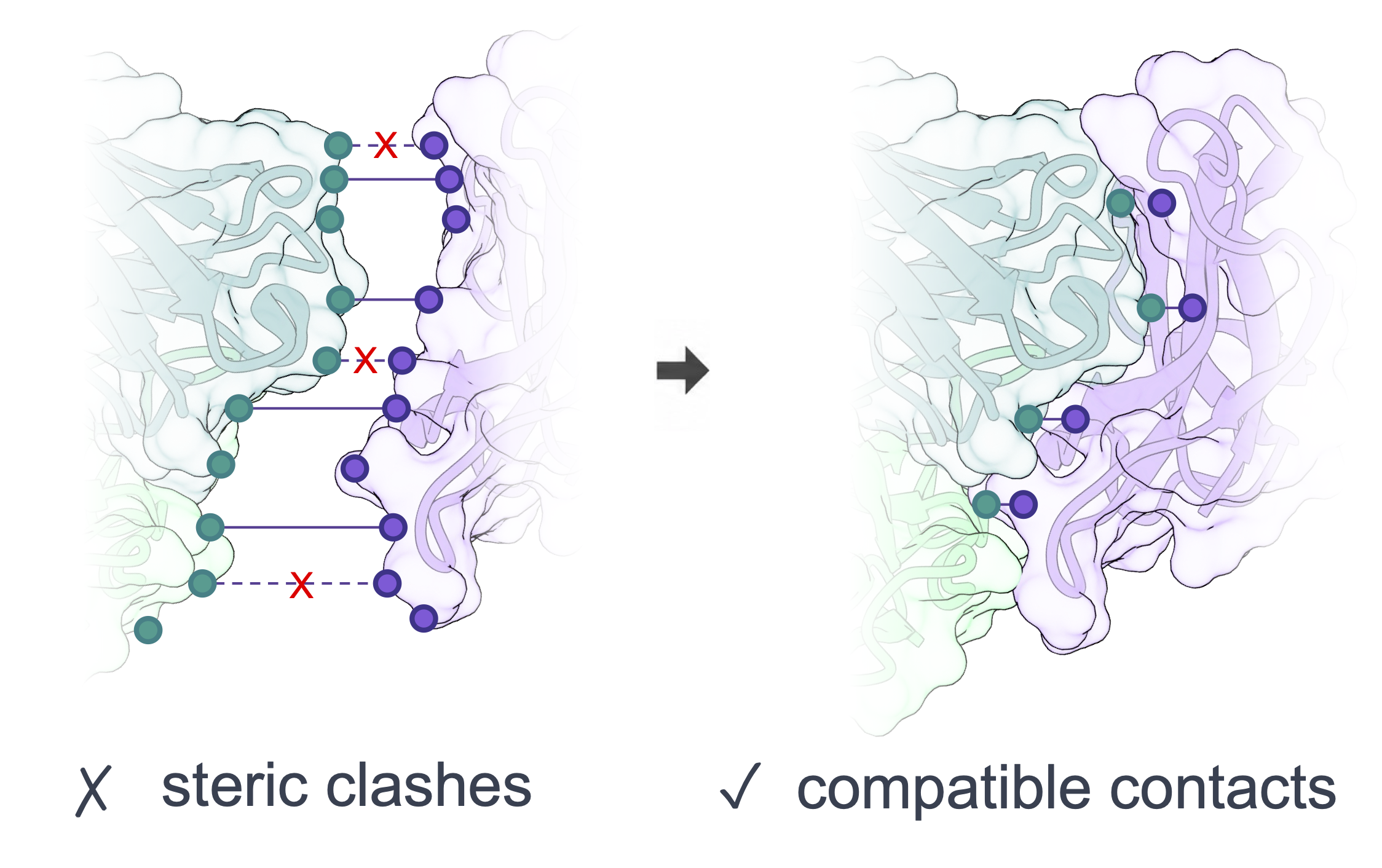
Shape-complementarity loss
Distance-only supervision such as RMSD cannot rule out steric clashes or interface gaps. OpenDDE adds a geometry-aware objective to diffusion training that scores predicted interfaces against native ones along three axes — token-level surface orientation, inter-chain spacing, and anti-clash contact quality — teaching the model the lock-and-key geometry that governs how biomolecules actually fit together.
One architecture for prediction and design
Structure prediction and de novo design are the same conditional-denoising problem. Known-target atom masks decide the mode: leave the mask empty and OpenDDE runs unconditional prediction; fix part of the system — an antigen backbone, a target pocket — and the same model becomes a design engine. No separate generative stack, just one end-to-end model.
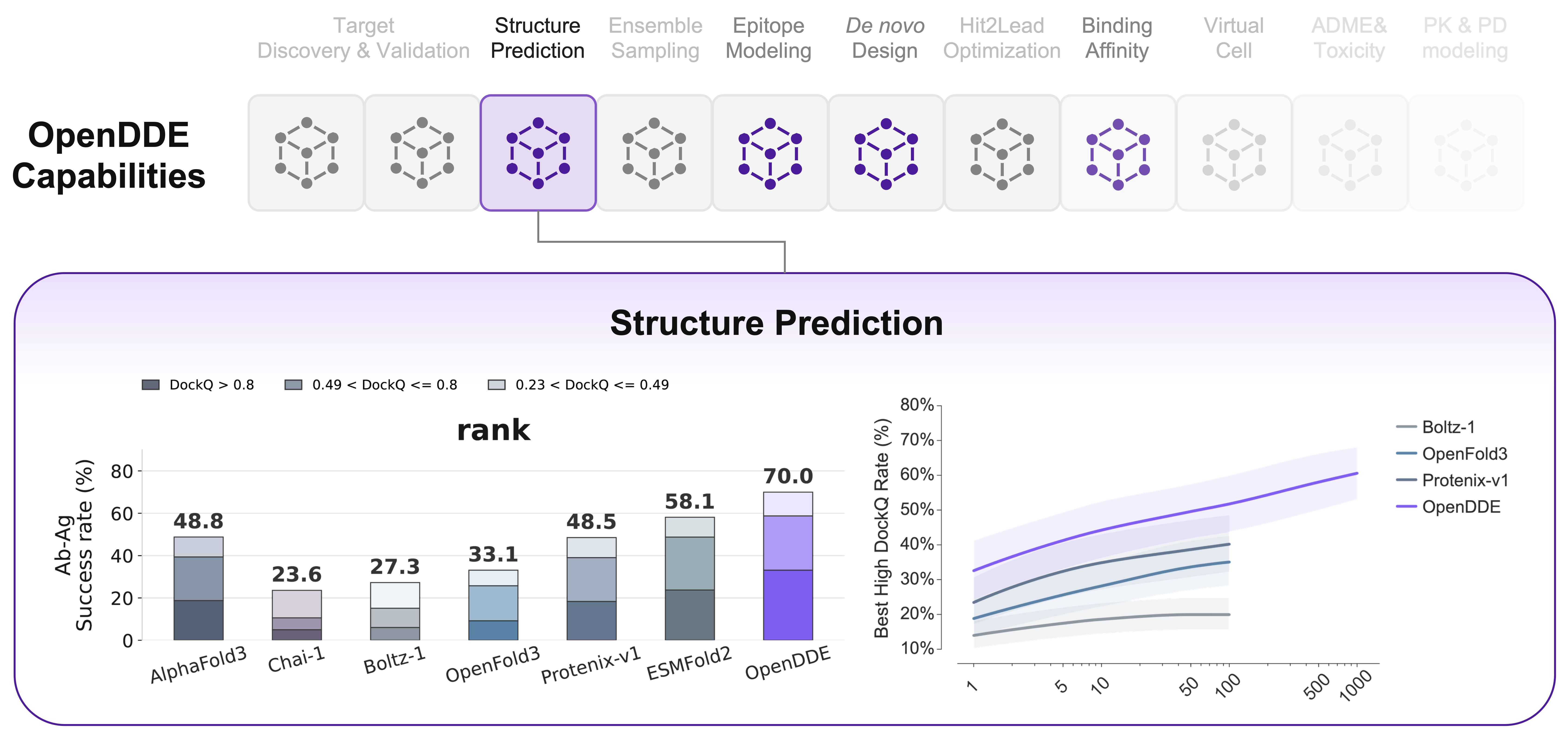
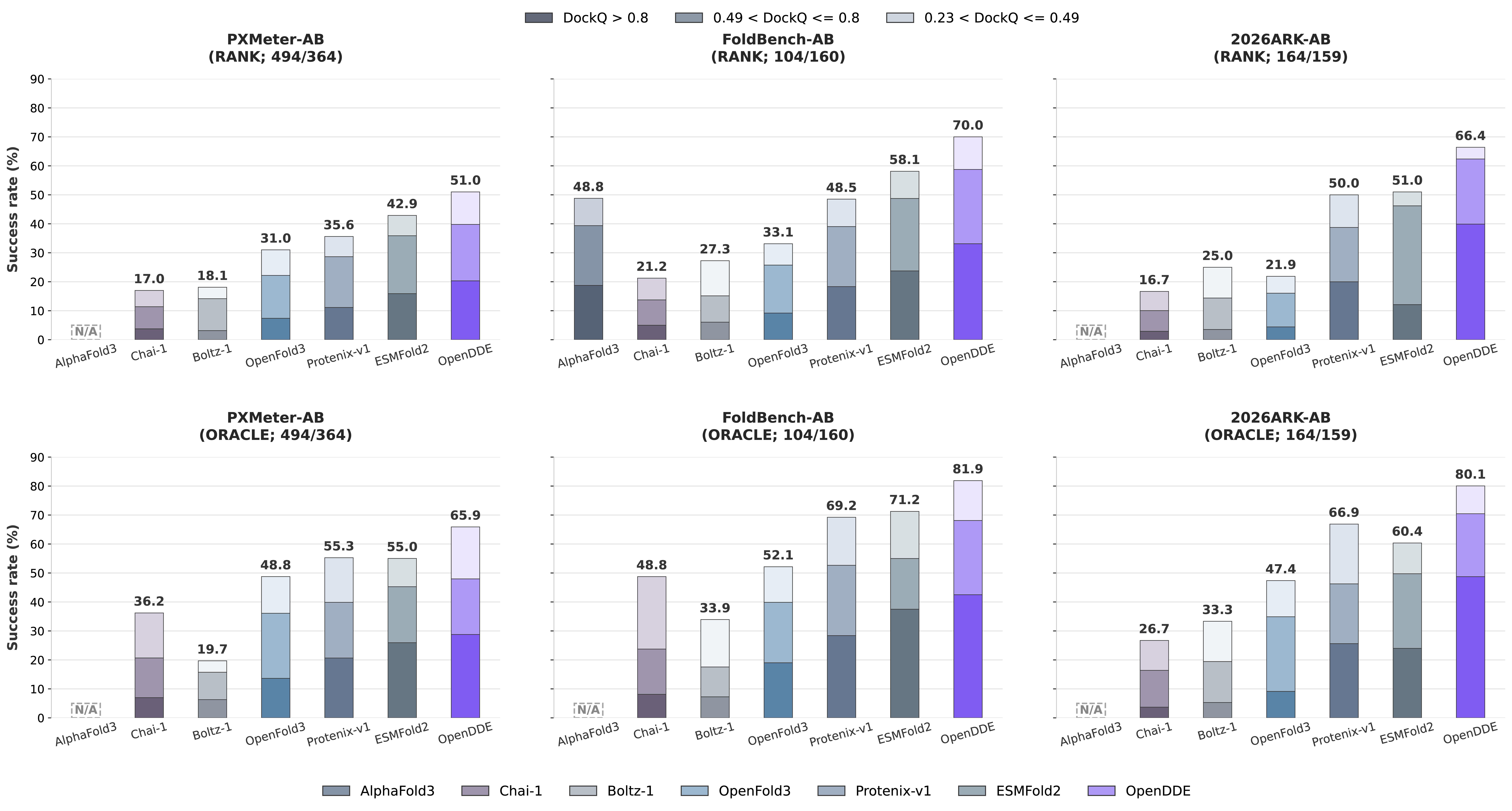
State of the art on antibody–antigen structure
Across three antibody–antigen benchmarks — PXMeter-AB, FoldBench-AB and 2026ARK-AB — OpenDDE leads every model tested on success rate, under both RANK (top-1) and ORACLE (best-of-N) selection, and across all DockQ quality tiers.
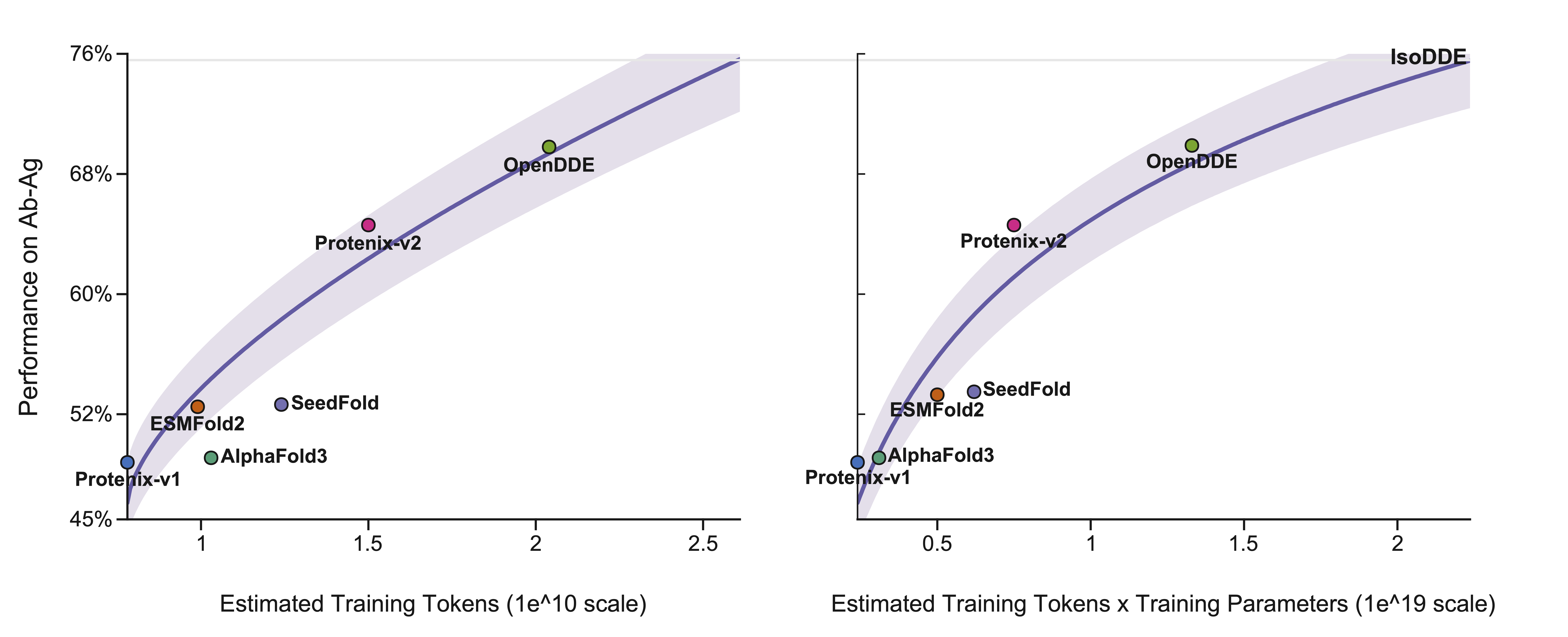
On the scaling frontier
Plotted against estimated training compute, OpenDDE sits on the efficient frontier of Ab-Ag performance — extracting more accuracy per token and per parameter than prior folding models, and pointing toward further gains as the recipe scales.
Built on open science
OpenDDE builds on ideas and components from the AlphaFold 3 ecosystem. If you use it in your work, please cite the software and the related research.